
La maladie de Willebrand
La maladie de Willebrand est une pathologie hémorragique héréditaire qui se traduit par une anomalie du facteur von Willebrand (VWF). Cette protéine sanguine participe au processus de l’hémostase, un phénomène permettant d’enrayer les saignements grâce à la formation de caillots sanguins. Découvrez les caractéristiques de cette maladie et les options thérapeutiques pour la traiter.
Qu’est-ce que la maladie de Willebrand ? 1-8
Décrite par le médecin finlandais Erik von Willebrand en 1926, la maladie de Willebrand (MW) est une pathologie génétique à risque hémorragique. Elle est causée par un déficit quantitatif et/ou défaut qualitatif du facteur Willebrand (VWF). Cette protéine sanguine est essentielle à l’hémostase, un ensemble de phénomènes physiologiques qui participent à l'arrêt des saignements.
Le facteur Willebrand se trouve dans le plasma, à l’intérieur des plaquettes – des cellules circulant dans le sang et contribuant à sa coagulation –, et sur les parois des vaisseaux sanguins. Il stimule l’agrégation et le maintien des plaquettes les unes aux autres et permet la formation d’un caillot sanguin solide appelé « caillot fibrino-plaquettaire », qui enraye les saignements. Lorsqu’il n’y a pas suffisamment de VWF dans le sang ou qu’il est défectueux, les plaquettes n’adhèrent pas à la paroi vasculaire de la région lésée. Il en résulte une prolongation anormale du saignement.
Parallèlement, le VWF est nécessaire au bon fonctionnement d’un autre facteur de coagulation, le facteur VIII : il le transporte dans le sang circulant, préservant sa durée de vie. La maladie de von Willebrand peut ainsi également entraîner un déficit modéré en facteur VIII, et donc amplifier les symptômes du trouble de la coagulation.
Il existe trois principaux types de maladie de Willebrand, dont la sévérité varie en fonction du degré de carence du facteur :
● La maladie de Willebrand de type 1 résulte d’un déficit quantitatif partiel en VWF. Ce dernier n’est pas altéré mais il est fabriqué en plus faible quantité que la normale, ou a une durée de vie plus courte, dans la circulation sanguine. Cette forme est considérée comme la plus fréquente et représente 50 à 75 % des cas.
●La maladie de Willebrand de type 2 découle d’un déficit qualitatif du VWF. Il se trouve en quantité normale ou peu diminuée dans la circulation sanguine mais est altéré dans sa structure. Cette forme de la maladie se subdivise, quant à elle, en plusieurs sous-types : 2A, 2B et 2M et 2N. Elle représente 20 à 45 % de tous les cas.
● La maladie de Willebrand de type 3 correspond à un déficit quantitatif sévère de VWF dans le sang. Il s’accompagne d’un taux très diminué de facteur VIII, équivalent à une hémophilie A légère à modérée. Cette forme sévère est néanmoins très rare, puisqu’elle affecte moins de 5 % des patients atteints par cette pathologie.
Combien de personnes sont atteintes de la maladie de von Willebrand ?
La prévalence de la maladie de Willebrand – soit le nombre de personnes malades au cours d'une période donnée –, est difficile à estimer. Les formes les plus modérées de la MW n’étant pas toujours accompagnées de saignements, seule une faible proportion d’entre elles sont diagnostiquées. Le nombre de patients qui nécessite un traitement au moins une fois dans leur vie oscille entre 1 sur 50 000 et 1 sur 8 500. En France, ce sont 2 506 patients qui ont été diagnostiqués avec la maladie de Willebrand, et référencés par le Réseau FranceCoag.
Comment la maladie de Willebrand est-elle transmise ? 1,2
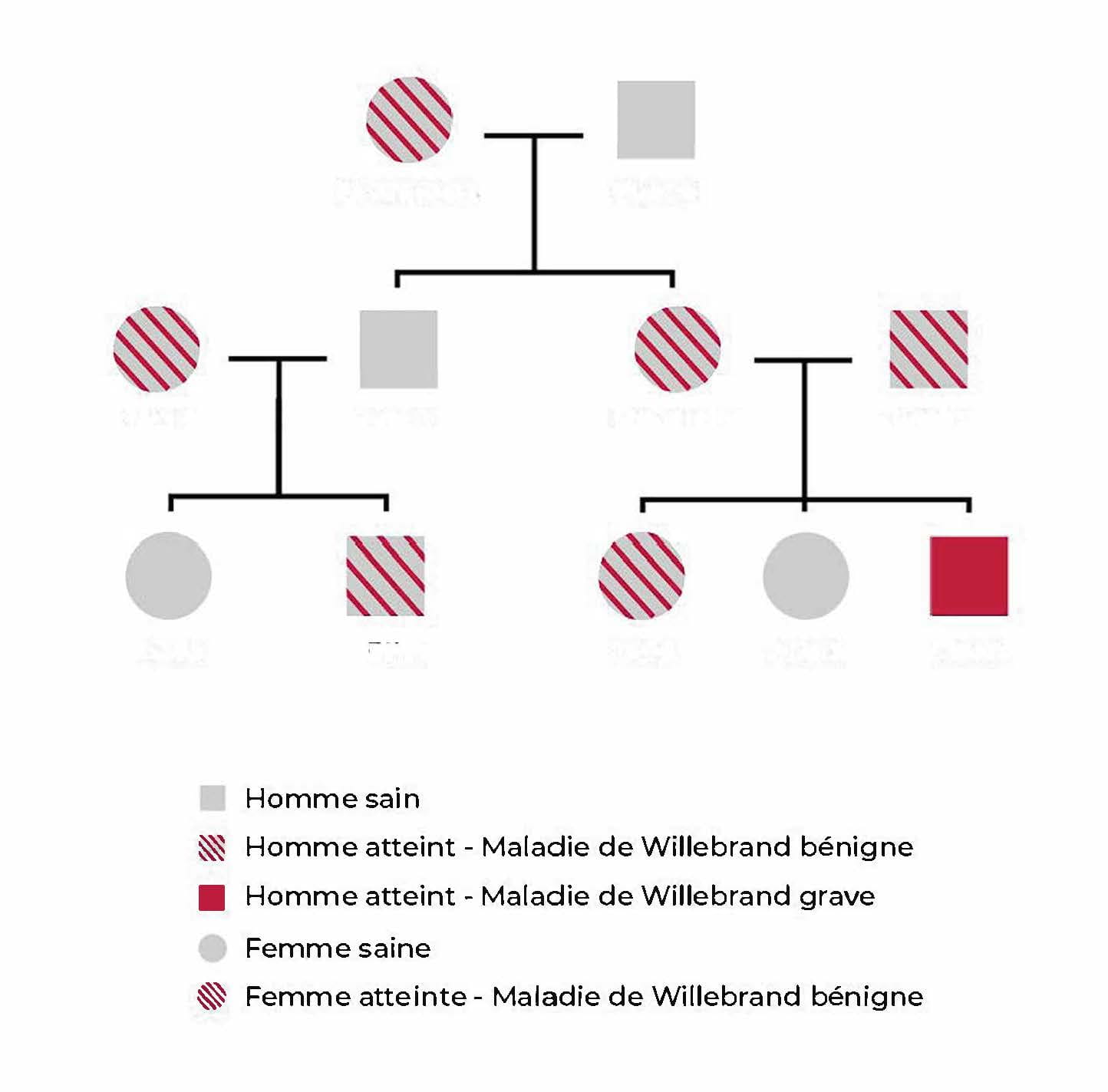
La maladie de Willebrand est causée par la mutation du gène VWF qui possède l’information permettant de fabriquer le facteur Willebrand. Ce gène est localisé sur le chromosome 12. La transmission est donc autosomique, c’est-à-dire non liée aux chromosomes sexuels (X ou Y). Elle affecte autant les hommes que les femmes, mais ces dernières présentent des symptômes plus notables en raison de leur vie gynécologique et notamment des risques hémorragiques liés aux règles abondantes, et des saignements prolongés après l’accouchement.
Elle peut être transmise héréditairement par l’un des deux parents porteurs du gène défectueux à l’enfant au moment de sa conception, et ce même s’il n’en manifeste aucun symptôme. Exceptionnellement, sa survenue peut être due à une mutation génétique non transmise par les parents : le chromosome 12 change au moment de la conception ou peu après. C'est ce que l'on appelle une mutation spontanée ou néomutation.
En cas de maladie de Willebrand de type 1 et la plupart des type 2, la transmission de la maladie est dominante : la personne malade possède une copie du gène défectueux. Elle aura un risque sur deux de transmettre la maladie à ses enfants. Dans le type 3, ainsi que quelques type 2 (2N et 2A), la transmission est récessive : la personne atteinte de la MW a reçu un exemplaire du gène muté de ses deux parents et aura un risque sur quatre de transmettre la maladie à ses enfants.
Quels en sont les symptômes ? 1,2
La plupart du temps, les personnes atteintes de la maladie de Willebrand sont asymptomatiques ou présentent des manifestations hémorragiques cutanéo-muqueuses. Autrement dit, des saignements qui se situent au niveau :
- de la peau ; avec des ecchymoses au moindre choc et des saignements prolongés résultant de petites coupures,
- des muqueuses ; avec des saignements du nez (épistaxis) et des gencives (gingivorragies), des saignements anormaux après une intervention chirurgicale (ex : une extraction dentaire).
Les femmes peuvent présenter des règles abondantes ou prolongées appelées ménorragies. La sévérité des signes hémorragiques de la maladie de Willebrand dépend du type de la maladie. En règle générale, le type 1 et le type 2 sont caractérisés par des symptômes légers à modérés. Cela dit, ils peuvent entraîner des épisodes hémorragiques graves en cas d’intervention chirurgicale, d’où l’intérêt de diagnostiquer la maladie. Les personnes atteintes de la Maladie de Willebrand de type 3 peuvent, quant à elles, présenter des saignements spontanés sans cause apparente, à l’intérieur des muscles (hématomes) et des articulations (hémarthroses). D’autres saignements – notamment au niveau du cerveau, du thorax ou de l’abdomen –, peuvent entraîner un risque vital s’ils ne sont pas rapidement pris en charge.
Comment est établi le diagnostic ? 1,2,7
La maladie de Willebrand est encore peu connue des médecins non spécialistes, rendant son diagnostic difficile. Elle est suspectée devant un patient qui présente des saignements exagérés ou inexpliqués ou dans le cadre d’une enquête familiale.
Trois paramètres sanguins sont dosés : le taux de facteur VIII, le taux de facteur Willebrand (plus précisément de l’antigène du facteur Willebrand) et la capacité du facteur Willebrand à se lier aux plaquettes (on parle ici d’activité du cofacteur de la ristocétine). Le comptage du nombre de plaquettes et la mesure du temps de saignement – temps que mettent les plaquettes à colmater une plaie cutanée minime à l’avant-bras –, sont également réalisés. D’autres tests plus spécialisés pourront être nécessaires pour déterminer le type ou le sous-type de la maladie de Willebrand.
Certains événements, tels qu’une infection ou une grossesse, augmentent de manière transitoire les taux de facteur Willebrand, pouvant entraîner des faux négatifs. Il est donc nécessaire que les examens sanguins soient reproduits, si la maladie est suspectée.
Quelles sont les options thérapeutiques ? 1, 2, 4
En dehors des accidents ou situations à risques hémorragiques (ex : intervention chirurgicale), de nombreuses personnes atteintes de la maladie de Willebrand ne requièrent pas de traitement. Les saignements mineurs se tarissent spontanément ou à l’aide de mesures simples, comme comprimer une petite plaie cutanée pendant quelques minutes. Cela dit, des traitements par agents anti-fibrinolytiques comme l’acide tranéxamique ou l’acide aminocaproïque peuvent aider à maîtriser les saignements au niveau des muqueuses – dans le nez, la bouche, les intestins ou l’utérus – en stabilisant les caillots, une fois qu’ils sont formés.
Dans certains cas toutefois, des traitements spécifiques peuvent corriger le déficit en facteur Willebrand (VWF). Ces traitements sont de deux types :
- La desmopressine est utilisée pour prévenir les épisodes de saignements lors de situations à risque ou pour les traiter. Elle permet de normaliser voire d’augmenter le taux du VWF dans le sang circulant en puisant le facteur Willebrand stocké dans l’organisme, notamment au niveau de la paroi des vaisseaux sanguins. Administrée par voie intraveineuse ou en inhalation intra-nasale, la desmopressine normalise la coagulation pendant quelques heures seulement.
- Les concentrés de facteur Willebrand sont produits à partir de plasma sanguin humain, fractionné pour en extraire le VWF et le facteur VIII, ou alors issu du génie génétique. Dans le premier cas, on parle de concentrés plasmatiques, et dans le second, de facteurs de coagulation recombinants. L’apport extérieur de ces facteurs de coagulation permet de normaliser la coagulation des malades de Willebrand. Ils sont administrés par voie intraveineuse aux patients non répondeurs à la desmopressine ; c’est-à-dire à tous les malades atteints du type 3, et à la plupart de ceux atteints du type 2. Ils sont également utilisés en cas d’hémorragie majeure ou de chirurgie importante chez tous les patients. Leur injection doit cependant être répétée tant que persiste le risque hémorragique, car leur durée d’action est limitée à quelques heures.
CSL Behring, engagé auprès des patients atteints de la maladie de Willebrand
Depuis plus d’un siècle, le laboratoire de biotechnologies CSL Behring a pour vocation de découvrir, développer et fournir des thérapies innovantes dérivées du plasma sanguin. Celui-ci permet de traiter des patients atteints de maladies rares et graves, telles que la maladie de Willebrand.
Pour contribuer à une meilleure prise en charge des patients atteints de maladies rares, le laboratoire estpartenaire des associations de patients telle que la Fédération Mondiale de l’Hémophilie. Le laboratoire CSL Behring a également développé une plateforme dédiée aux troubles de la coagulation:Hémophilink. L’objectif : accompagner au quotidien les patients et leurs proches.
***
Références :
1. Tout sur la maladie de von Willebrand... à l’intention des personnes atteintes de la maladie de von Willebrand et de leurs proches. Brochure de la Société Canadienne de l’hémophilie. Décembre 2011. Consultée sur https://www.hemophilia.ca/wp-content/uploads/2018/05/Tout-sur-la-maladie-de-von-Willebrand-2011.pdf
2. La maladie de Willebrand. Brochure de l'Encyclopédie Orphanet Grand Public. Novembre 2006. Consultée sur www.orpha.net/data/patho/Pub/fr/Willebrand-FRfrPub3497v02.pdf
3. Site Internet de l’Association Française de l’Hémophilie, Section : Je m’informe. Page : Qu’est-ce que la maladie de Willebrand ? consultée le 22/11/2019 https://afh.asso.fr/je-minforme/comprendre-les-maladies-hemorragiques/maladie-de-willebrand/quest-ce-que-la-maladie-de-willebrand/
4. Site Internet de l’Association Française de l’Hémophilie, Section : Je m’informe. Page : La maladie de Willebrand de type 1, consultée le 22/11/2019 https://afh.asso.fr/je-minforme/comprendre-les-maladies-hemorragiques/maladie-de-willebrand/la-maladie-de-willebrand-de-type-1/
5. Site Internet de l’Association Française de l’Hémophilie, Section : Je m’informe. Page : La maladie de Willebrand de type 2, consultée le 22/11/2019 https://afh.asso.fr/je-minforme/comprendre-les-maladies-hemorragiques/maladie-de-willebrand/la-maladie-de-willebrand-de-type-2/
6. Site Internet Le manuel MDS, version pour le grand public. Page : La maladie de Von Willebrand : https://www.msdmanuals.com/fr/accueil/troubles-du-sang/maladies-des-plaquettes/maladie-de-von-willebrand, consultée le 07/02/2020
7. Site Internet Le manuel MDS, version pour les professionnels de santé. Page : La maladie de Von Willebrand : https://www.msdmanuals.com/fr/professional/h%C3%A9matologie-et-oncologie/thrombop%C3%A9nie-et-dysfonctionnement-des-plaquettes/maladie-de-von-willebrand, consultée le 07/02/2020
8. Site Internet du portail et serveur d'informations dédié aux maladies rares et aux médicaments orphelins en libre accès pour tous publics Orphanet. Section : Maladies rares. Page : Maladie de von Willebrand https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=FR&Expert=903, consultée le 10/02/2020
9. Site internet du Réseau FranceCoag, Page Statistiques nationales sur la Maladie de Willebrand, https://www.francecoag.org/SiteWebPublic/public/stats/stats_page.jsp?stat4=on, consultée le 23/03/20